2024.01.29
發表人:洪右真/研究員
孩子確診聽損後,除了張羅輔具和準備進入療育課程外,常常還得苦惱是否應該打破砂鍋問到底,加碼進行基因檢測來釐清孩子聽損的成因。但是,基因檢測真的有意義嗎?我們家又沒有人有聽損,小孩怎麼可能是遺傳性聽損呢?到底甚麼是聽損基因?知道基因檢測結果又能做什麼?不妨就先從這篇文章開始,讓我們透過常見聽損基因的介紹、聽損基因檢測用處的說明,來替各位解答疑惑。
超過半數以上的聽損是基因變異導致
聽力損失為最常見的兒童感官缺陷,依據2022年國民健康署年報提供之數據計算,每1,000位胎兒中約有5名寶寶患有類型程度不一的先天性聽力損失(國民健康署,2022),其中約有一半是由基因變異引起。基因是攜帶遺傳訊息的基本單位,每個人身體裡大約有20,000到25,000個基因,基因組成DNA,是父母傳給子女的遺傳單位。
在我們的身體裡,基因都是成對存在的。一個來自媽媽,一個來自爸爸。一般情況下,這對基因幾乎相同,但有的時候,一個基因裡可能會發生一些改變,而如果剛好這個基因是負責耳朵相關的工作,就可能會導致聽力問題。現今已發現有超過100種基因與聽覺功能息息相關,其中有一些基因若表現異常便會造成聽力損失。
七成遺傳性聽損屬非症候群型
以圖一說明,顯示了約有5成先天性聽損的成因與基因相關,屬於遺傳性聽損,其中大概有7成的聽損兒童屬非症候群型(Non-syndromic),也就是除了聽損以外沒有其他的併發症狀(Shearer et al., 1999)。另外大約有3成的聽損兒會伴隨其他器官異常和病症,屬於症候群型(Syndromic)。目前已知合併聽損的症候群就超過400種,常見的包括彭德萊症候群(Pendred syndrome)、尤塞氏症候群(Usher syndrome)和瓦登伯格症候群(Waardenburg syndrome)等(Venkatesh et al., 2015)。不論何種類型,聽力損失皆可能發生於單側或雙側耳朵,聽損程度可輕可重,有的屬於穩定型,有些則是漸進型,也就是聽損程度會隨著時間變重。
八成以上遺傳性聽損兒的父母聽力正常
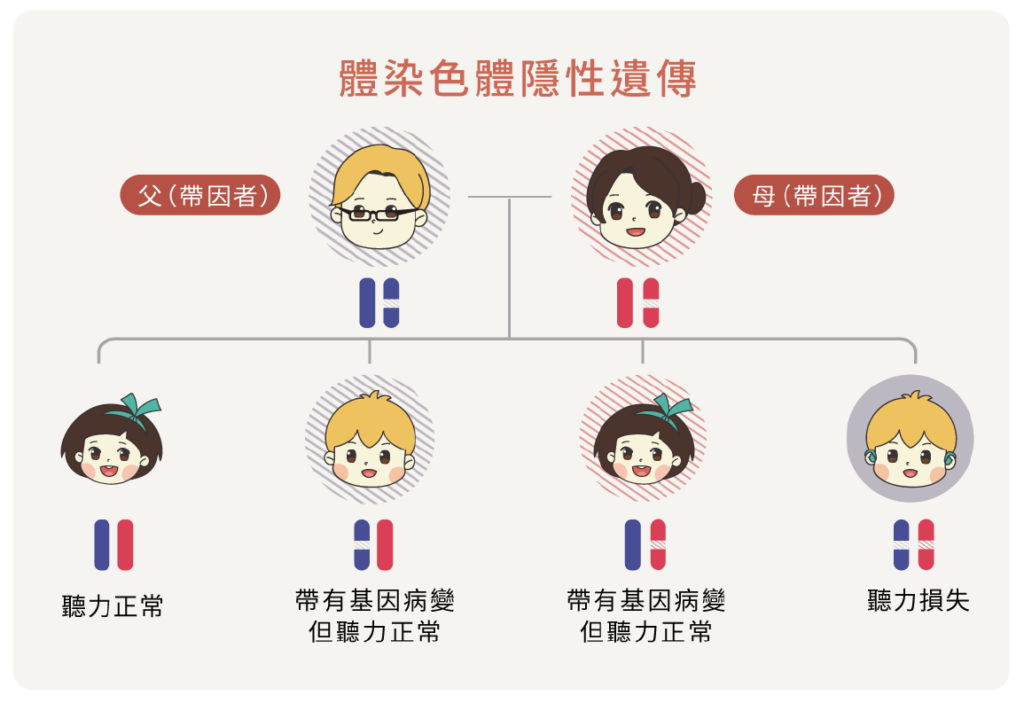
聽損遺傳模式可分為體染色體隱性遺傳、體染色體顯性遺傳、性聯遺傳和粒線體遺傳。其中非症候群型聽損中以高達80%的體染色體隱性遺傳最為常見,這也就是為什麼父母或是家族中都沒有人聽損,但孩子還是有可能透過遺傳成為聽損兒。因為即便父母的聽力正常,但是因各自帶有變異基因,也就是「帶因者」,其子女便有25%的機率會成為聽損者、50%的機率成為聽損帶因者,和25%的機率不會成為聽損帶因者。目前已知有超過60個基因會導致體染色體隱性非症候群的聽損,其中臨床上最常見的是GJB2和SLC26A4的基因變異(Brownstein et al., 2012)。
體染色體顯性遺傳則為父母有一人為聽損患者,則其子女便有一半的機率繼承聽損基因並成為患者。目前已知有超過30個基因會導致體染色體顯性非症候群的聽損。性聯遺傳和粒線體遺傳相形之下較為少見,其中較為常見的是粒線體12S rRNA基因異常,屬於母系遺傳。
台灣常見的聽損基因
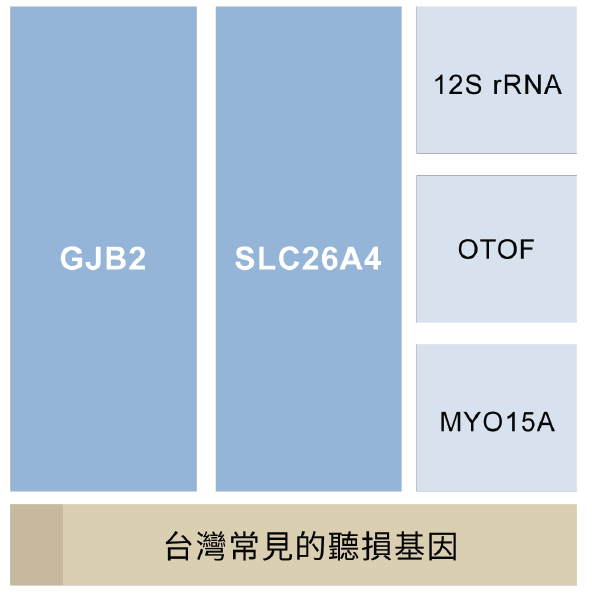
雖然已知的聽損基因就超過百種,但根據2008年國內研究指出,台灣最常見的三種聽損基因為GJB2, SLC26A4 和粒線體12S rRNA,分別佔了21.7%,14.4%,以及 3.8%(Wu et al., 2008)。然而,隨著基因檢測技術的推進,近幾年的研究進一步顯示,除了GJB2和SLC26A4仍為前兩大國人最常見聽損基因外,OTOF、MYO15A則和已知的12S rRNA並列為另三種常見的聽損基因(Wu et al., 2019; Lee, et al., 2023)。考量現今大部分醫療院所的基礎基因篩檢項目仍以GJB2、SLC26A4和粒線體12S rRNA為主,本篇將針對這三種常見基因進行說明:
GJB2
在非症候群中,最常見的基因變異出現在Gap Junction Beta 2基因(GJB2)中,佔了高達50%隱性遺傳聽損的病例,也就是,有接近20%的先天性聽損與GJB2有關(Estivill et al., 1998; Kelley et al., 1998)。GJB2基因的變異會讓耳蝸中的鉀離子不平衡,讓內耳無法正常運作,導致聽損。在台灣,GJB2 基因較為常見的變異分別為 c.109G>A (p.V37I) 與c.235delC(Wu et al., 2008)。
這串令人滿頭霧水的字母和數字到底代表甚麼意思?其實它們通常是在描述基因變異的狀況。能夠解讀這些字串,讓基因名稱不再像是無字天書,不僅能理解基因變異的狀況,也讓人更容易記憶完整的基因名稱。未來與專業人員討論病史時就可完整提供資訊,讓聽語療育服務輸送更精確順暢。就以GJB2 c.109G>A (p.V37I) 來說:
l c.109G>A:c.是coding DNA reference sequence (DNA編碼參考序列)的縮寫,用來描述突變基因的位置;109是指變化產生的位置,原本應該是G核酸的位置,卻被A核酸取代了。可以想像成打印編碼的過程中把「G」打成了「A」了。
l (p.V37I):p.代表蛋白質(protein),用來表示基因變異導致的蛋白質變化。原本位於蛋白質37位置的「V」(valine/纈氨酸)變成了「I」(isoleucine/異白胺酸)。
而GJB2 c.235delC則代表GJB2基因的DNA序列中,位置235的「C」(cytosine/胞嘧啶)被刪除,這邊del便是英文刪除(delete)的縮寫。
國內有相當多針對GJB2基因的研究,發現該基因變異會導致不同程度的損,可能為先天性,但也有可能為遲發型聽損,也就是出生後才出現聽損,且聽力狀況可能會隨著年紀逐漸加重。
若隨機檢視本會25名帶有GJB2 c.109G>A (p.V37I) 變異的聽損兒,有19位孩子(76%)出生時即因未通過新生兒聽力篩檢,進而確診聽損,但仍有6位孩子(24%)單耳或雙耳通過新生兒聽力篩檢。其中2名孩子出生時為聽力正常,但於約三歲時因家人觀察到其發展較慢或發音不標準而進行專業評估時發現聽損。聽損程度從極輕度到極重度皆有,但以輕度聽損比例最高(52%),接續為中度(36%)、極輕度(8%)和極重度(4%)聽損。
因仍有遲發型聽損的可能性,因此即便通過新生兒聽篩,還是應該密切注意孩子的聽能表現,若對於孩子聽語發展狀況有所擔憂,則可至醫療院所進行評估或檢查。
SLC26A4
SLC26A4為第二種常見的基因變異,發生率僅次於GJB2(Albert et al., 2006)。國內研究顯示約有15~20%的聽損兒帶有此變異基因,會讓維持耳蝸內淋巴液電解質濃度和甲狀腺中離子輸送的功能異常。SLC26A4中的SLC指的便是溶質載體(Solute Carrier),而SLC26A4代表了溶質載體家族中的一種特定的蛋白質,負責在細胞膜上運輸物質。SLC26A4可引起症候型和非症候型的聽力損失:
l 症候型:彭德萊症候群(Pendred syndrome),特徵為先天性感音神經性聽損、甲狀腺腫大和有時會伴隨平衡問題
l 非症候型:DFNB4孤立性聽損,為SLC26A4變異引起的聽力損失,沒有伴隨其他症狀
因為內耳結構異常,上述兩者也常合併前庭導水管擴大症(enlarged vestibular aqueduct syndrome,簡稱EVA),導致中度至重度的波動型聽力損失,聽力狀況時好時壞。許多研究顯示,聽力波動常會出現在頭部遭受撞擊或周遭空氣壓力遽變(如潛水活動)後發生(National Institutes of Health [NIH], 2017)。值得注意的是,前庭導水管擴大症導致的聽損不見得會在出生當下出現,有時會在兒童時期甚至更晚才發生,因此孩子還是有可能會通過新生兒聽力篩檢。
12S rRNA
12S rRNA代表一種核糖體,在蛋白質合成過程扮演重要的角色。負責編碼12S rRNA的粒線體DNA變異時便會容易影響內耳正常運作,導致聽力損失。尤其變異位點若為m.1555A>G,則應注意避免使用耳毒性抗生素,如胺基酸甘醣體類(Aminoglycosides)藥物,以避免聽力進一步惡化。
基因檢測,對聽力發展多一分掌握
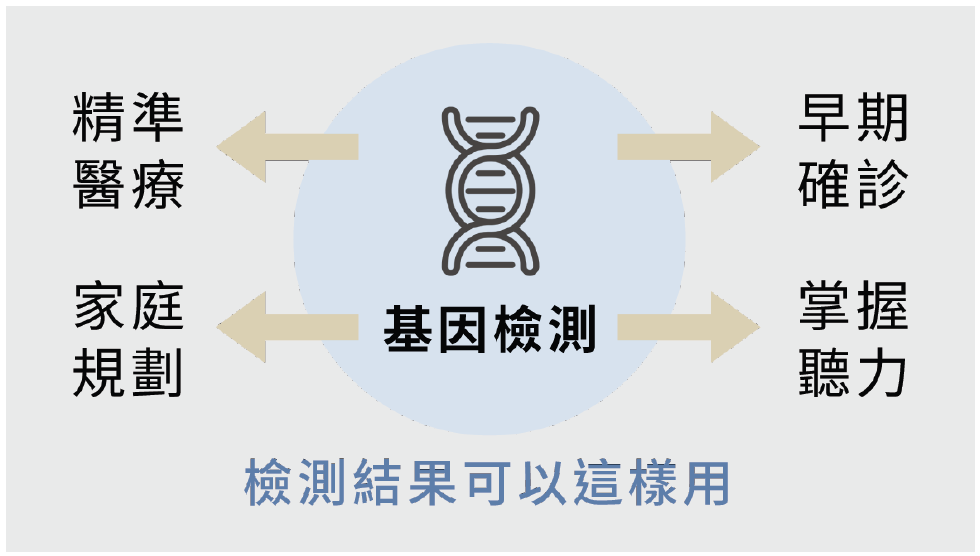
約有3成感音神經性聽損兒帶有上述其中一種的基因變異。家長可以依據家族病史、自身和小孩狀況,向醫生諮詢是否有進行聽損基因檢查的必要。從聽力保健角度來看,基因檢查的結果有時能讓醫療處置更精準,也能讓父母主動出擊,掌握孩子未來聽力的走向:
- 早期確診:除了聽損,還有其他功能異常嗎?有些聽力損失屬於症候群類型,透過基因檢測將能進一步找出其他可能的病症,如彭德萊症候群就會合併甲狀腺功能異常。
- 有意識避免聽損惡化:不管是波動型或漸進型聽損孩子的父母都可透過日常生活中的觀察或是檢測(如雅文檢測音)有效掌握孩子的聽力變化。此外,也應有意識避免會讓聽力惡化的狀況,如:粒線體基因變異的孩子不能服用耳毒性藥物,或是前庭導水管擴大症的孩子應避免讓頭部受到撞擊。
- 讓治療選擇更有依據:GJB2、SLC26A4和粒線體12S rRNA基因變異皆會導致內耳功能無法正常運作,導致聽損,其病理位置皆位於耳蝸。因此,根據研究結果,醫師學者普遍對於這類患者植入人工電子耳後的效益保持相對樂觀的態度。
- 家庭規劃:若父母有生育打算,可透過檢查結果進行生孕諮詢,評估遺傳性聽損會發生的機率。
雖然從統計資料可知,有一半兒童的聽損成因來自遺傳因素,但由於有八成以上遺傳性聽損孩子的父母通常聽力沒有異狀,故容易讓人忽略或無法接受孩子為遺傳性聽損的可能。每一種檢測的做與不做皆有其考量,沒有對錯,聽損基因檢測也是一樣的道理。雖然基因檢測可能會讓聽損成因真相大白,但也有可能會導致父母的焦慮、內疚和產生對未來的不安感。最後,建議父母可以先整理好自己的想法,再和醫師討論諮詢,權衡基因檢測的利與弊。
(全文刊登於 NO.44雅文聽語期刊 遺傳性聽力損失)
參考文獻
國民健康署年報(2022)。檢自(2023.10.18):https://health99.hpa.gov.tw/m7958
Albert, S., Blons, H., Jonard, L., Feldmann, D., Chauvin, P., Loundon, N., Sergent-Allaoui, A., Houang, M., Joannard, A., Schmerber, S., Delobel, B., Leman, J., Journel, H., Catros, H., Dollfus, H., Eliot, M. M., David, A., Calais, C., Drouin-Garraud, V., Obstoy, M. F., Tran Ba Huy, P., Lacombe, D., Duriez, F., Francannet, C., Bitoun, P., Petit, C., Garabédian, E. N., Couderc, R., Marlin, S., & Denoyelle, F. (2006). SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. European. Journal of Human Genetics, 14(6), 773-779.
Brownstein Z., Bhonker Y., & Avraham K.B. (2012). High-throughput sequencing to decipher the genetic heterogeneity of deafness. Genome Biology 13, 245–255.
Estivill, X., Fortina, P., Surrey, S., Rabionet, R., Melchionda, S., D’Agruma, L., Mansfield, E., Rappaport, E., Govea, N.,Milà, M., Zelante, L., & Gasparini, P. (1998). Connexin-26 mutations in sporadic and inherited sensorineural deafness. The Lancet, 351(9100), 394-398.
Kelley, P. M., Harris, D. J., Comer, B. C., Askew, J. W., Fowler, T., Smith, S. D., & Kimberling, W. (1998). Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. The American Journal of Human Genetics, 62(4), 792-799.
Lee, Y. H., Tsai, C. Y., Lu, Y. S., Lin, P. H., Chiang, Y. T., Yang, T. H., Hsu, J. S., Hsu, C. J., Chen, P. L., Liu, T. C., & Wu, C. C. (2023). Revisiting Genetic Epidemiology with a Refined Targeted Gene Panel for Hereditary Hearing Impairment in the Taiwanese Population. Genes, 14(4), 880. https://doi.org/10.3390/genes14040880
National Institutes of Health. (2017). Enlarged Vestibular Aqueducts and Childhood Hearing Loss. Retrieved October 03, 2023, from https://www.nidcd.nih.gov/health/enlarged-vestibular-aqueducts-and-childhood-hearing-loss#:~:text=To%20reduce%20the%20likelihood%20of,to%20barotrauma%20(extreme%2C%20rapid%20changes
Shearer, A. E., Hildebrand, M. S., Schaefer, A. M., & Smith, R. J. H. (1999). Genetic Hearing Loss Overview. In M. P. Adam (Eds.) et. al., GeneReviews®. University of Washington, Seattle. [Updated 2023 Jun 29] Available from: https://www.ncbi.nlm.nih.gov/books/NBK1434/
Sheffield, A. M., & Smith, R. J. (2019). The epidemiology of deafness. Cold Spring Harbor perspectives in medicine, 9(9): a033258.
Venkatesh, M. D., Moorchung, N., & Puri, B. (2015). Genetics of non syndromic hearing loss. Medical Journal Armed Forces India, 71(4), 363-368.
Wu C. C., Chen P. J., Chiu Y. H., Lu Y.C., Wu M. C., & Hsu C. J. (2008). Prospective mutation screening of three common deafness genes in a large Taiwanese Cohort with idiopathic bilateral sensorineural hearing impairment reveals a difference in the results between families from hospitals and those from rehabilitation facilities. Audiology and Neurotolog.13(3):172-81. doi: 10.1159/000112425.
Wu, C. C., Tsai, C. Y., Lin, Y. H., Chen, P. Y., Lin, P. H., Cheng, Y. F., Wu, C. M., Lin, Y. H., Lee, C. Y., Erdenechuluun, J., Liu, T. C., Chen, P. L., & Hsu, C. J. (2019). Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes, 10(10), 772. https://doi.org/10.3390/genes10100772